宏基因组(Metagenomics),也称元基因组,利用新一代高通量测序技术( NGS)以特定环境下微生物群体基因组为研究对象,在分析微生物多样性、种群结构、进化关系的基础上,可进一步探究微生物群体功能活性、相互协作关系及与环境之间的关系,发掘潜在的生物学意义。与传统微生物研究方法相比,宏基因组测序技术规避了绝大部分微生物不能培养、痕量菌无法检测的缺点,因此近年来在环境微生物学研究中得到了广泛应用。
技术优势
- 通量高,拥有标准化实验室和高通量测序平台,数据库可靠
- 可检测不可培养物种,可检测痕量微生物
- 专业的生物信息团队,可以满足个性化的生物信息分析要求
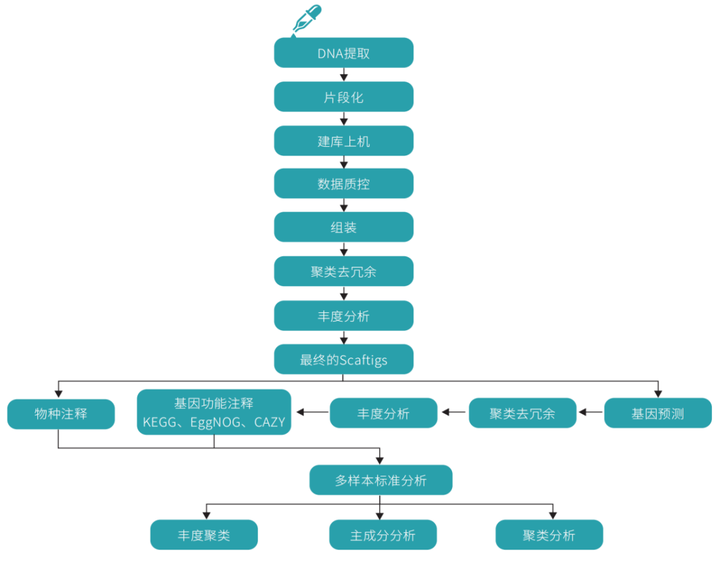
技术路线
技术参数
样本要求
土壤:10 g/sample
粪便:3-5 g/sample
血液:10 mL/sample
污泥/沉积物:5-10 g/sample
DNA:总量≥1 ug,浓度≥100 ng/uL
检测平台
测序平台:Illu mina Nova Seq 6000
测序方法:PE150
测序深度:6G/10G/12G
常规项目周期
实验检测:35个自然日(从样本质检合格及该批次首付款到位的次日起)
数据分析:7个自然日
应用方向
1.医学领域:代谢病研究、肿瘤癌症研究等
2.畜牧领域:肠道、瘤胃(如产甲烷菌类群)与动物健康/营养消化研究等
3.农业领域:根据微生物与植物互作、农业耕作/施肥处理与土壤微生物群落研究等
4.环境领域:雾霾处理、污水治理、石油降解、酸性矿水处理及海洋环境研究等
5.生物能源:特殊功能的菌株、基因挖掘、工程菌的开发研究
6.特殊极端环境:极端环境条件下的微生物类群研究
案例分析

宏基因组和代谢组分析揭示肠道菌群明显的结直肠癌阶段性特异表型
研究对象:人
期刊:Nature Medicine
影响因子:30.641
时间:2019年
研究背景
大多数散发性结直肠癌的发生是多步过程,首先形成息肉样腺瘤,然后发展为粘膜内癌,最后发展为恶性肿瘤,其形成需要历经几十年的时间,早期癌症的发现和内镜切除是癌症控制有效手段,肠道菌群与结直肠癌的发生有密切关系。
研究结果
基于宏基因组和代谢组研究平台,对来自大队列CRC中的616名和406名样本分别进行了粪便宏基因组和代谢组学研究,获得不同阶段(MP,S0,Sl /SIl,II/SIV,HS)病例特异性表型的微生物(Atopobiumparvulum和B.wadsworthia等)和代谢(氨基酸和胆汁酸等)标志物;且进一步探索个体直肠癌患者的肠道微生物组与肿瘤与特征之间的关系。
结果展示
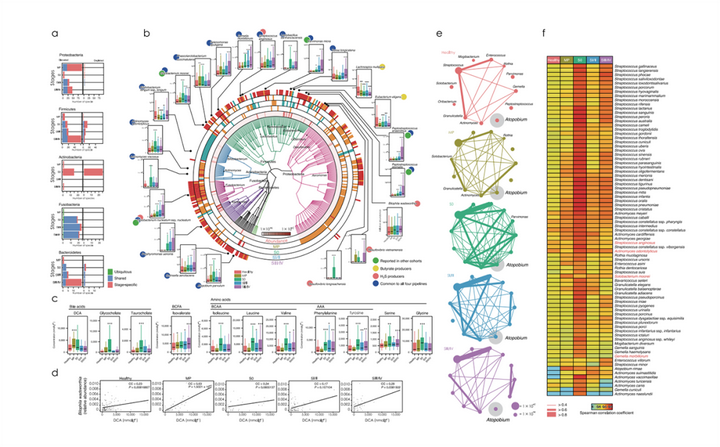
(1)宏基因组和代谢组分析
宏基因组分析首先发现Bacteroides和Prevotella是所有受试者和健康对照中变化最大的菌。进一步的研究还确定了与CRC关联的新物种,其中Colinsella aerofaciens,Dorealongicatena,Porphyro-monasuenonis和Streptococcus anginosus等在III/IV中显著升高。代谢组分析发现,大肠中主要能源物质丙酸盐和丁酸盐是含量最高的两种代谢物,二氢尿嘧啶和尿素也存在较大差异。与健康对照组相比,MP中DCA(脱氧胆酸盐)浓度显著升高;SO中甘氨胆酸盐和牛磺胆酸盐浓度显著升高;支链氨基酸,苯丙氨酸,酪氨酸,甘氨酸,丝氨酸浓度也升高。
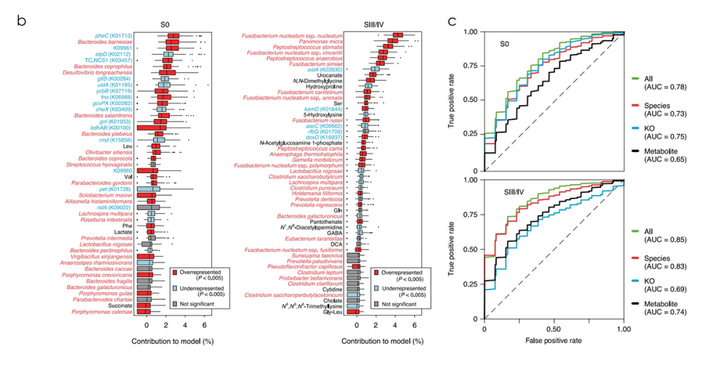
(2)功能分析和标志物筛选
基于KEGG数据库筛选出1243个同源基因(KO基因)在至少一个阶段显著升高,96个KO基因在至少一个阶段显著降低。在差异最显著的通路中,芳香族氨基酸代谢和产硫化物的通路与CRC相关。参与苯丙氨酸和酪氨酸合成的基因显著升高,其中pheC被确定为区分S0患者和健康对照的得分最高标志物。通过构建随机森林和LASSO逻辑回归模型来区分健康对照与SO和SIII/IV。在S0分类中,特征值是包括编码环己二烯基脱水酶的pheC的KO基因;在SIII/IV分类中,特征值为已被确认为CRC标志物的P.micraP.stomatis和F.nucleatum等口腔厌氧菌。
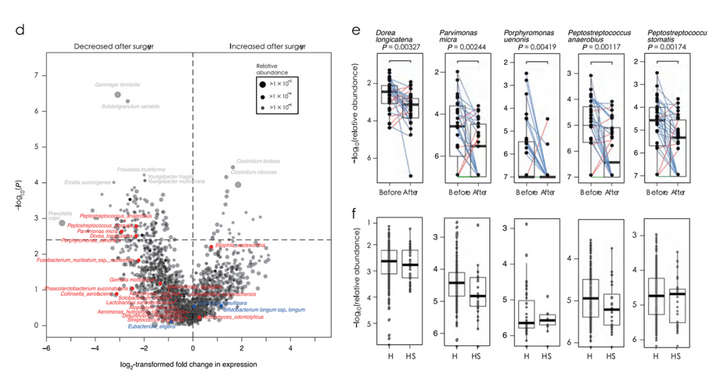
(3)结果验证和潜在机制
对28名CRC患者(SI /II和SIII /IV)术前和术后1年的粪便样品进行宏基因组测序。结果表明,肿瘤切除后P.stomatis,P.anaerobius和P.uenonis的相对丰度降低。
参考文献
文献下载链接:
https://pan.baidu.com/s/1bcWuKYjVHVH9mdaNxMI0VQ
提取码:0000